
 |
Darin R. Rokyta, PhD Assistant Professor Lab: 4070 King Life Sciences Office: 4058 King Life Sciences Contact | ||||
|
Below are a few examples of the types of research going on in the Rokyta lab. Adaptation TheoryCan any general predictions be made about an adapting population or the genetic changes underlying adaptation? Perhaps. Most mutations reduce fitness, so beneficial mutations, the raw material of adaptation, are rare. Rare events tend to have regular, predictable statistical properties (much like average events). Using this idea, we can derive predictions about short-term adaptive evolution. For example, the figure below shows the predictions for a single step in adaptation as a function of the parameter kappa, which describes the shape of the distribution of the effects of beneficial mutations. The heights of the vertical bars give the probability that natural selection will choose that particular mutation, where the best mutation has rank 1, the second best has rank 2, and so on. The predictions depend critically on kappa, which must be estimated empirically (see below). 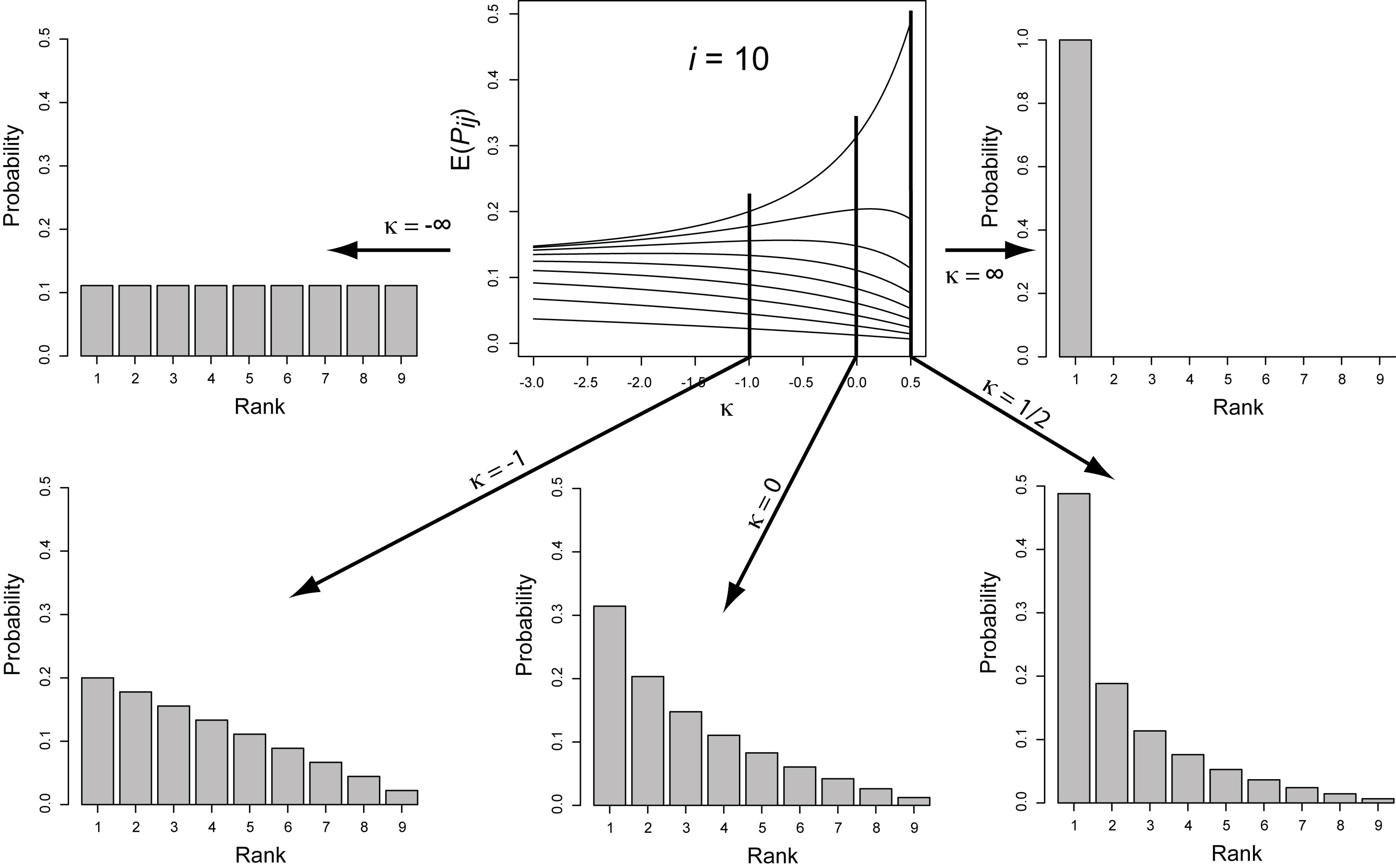 The details of the above model can be found in Joyce et al. (2008) in the publications section. Using this model, we have been able to characterize short-term adaptation for populations of moderate size under strong selection. We are interested in relaxing these two assumptions and extending the results to long-term adaptation. Experimental Viral EvolutionMuch of our experimental work is driven by adaptation theory. In some cases, we directly test theoretical predictions or the assumptions that form the basis of the models. Other work is meant to provide guidance for future theoretical investigations. For an example of how we can test model predictions, see Rokyta et al. (2005) in the publications section. Below is a figure showing how we can test model assumptions. Much previous adaptation theory has entailed the assumption that the effects of beneficial mutations follow an exponential distribution. Theoretical considerations actually suggest that the exponential is but one of three types of possible distributions. A statistical test for distinguishing among these possibilities is described by Beisel et al. (2007), listed in the publications section. 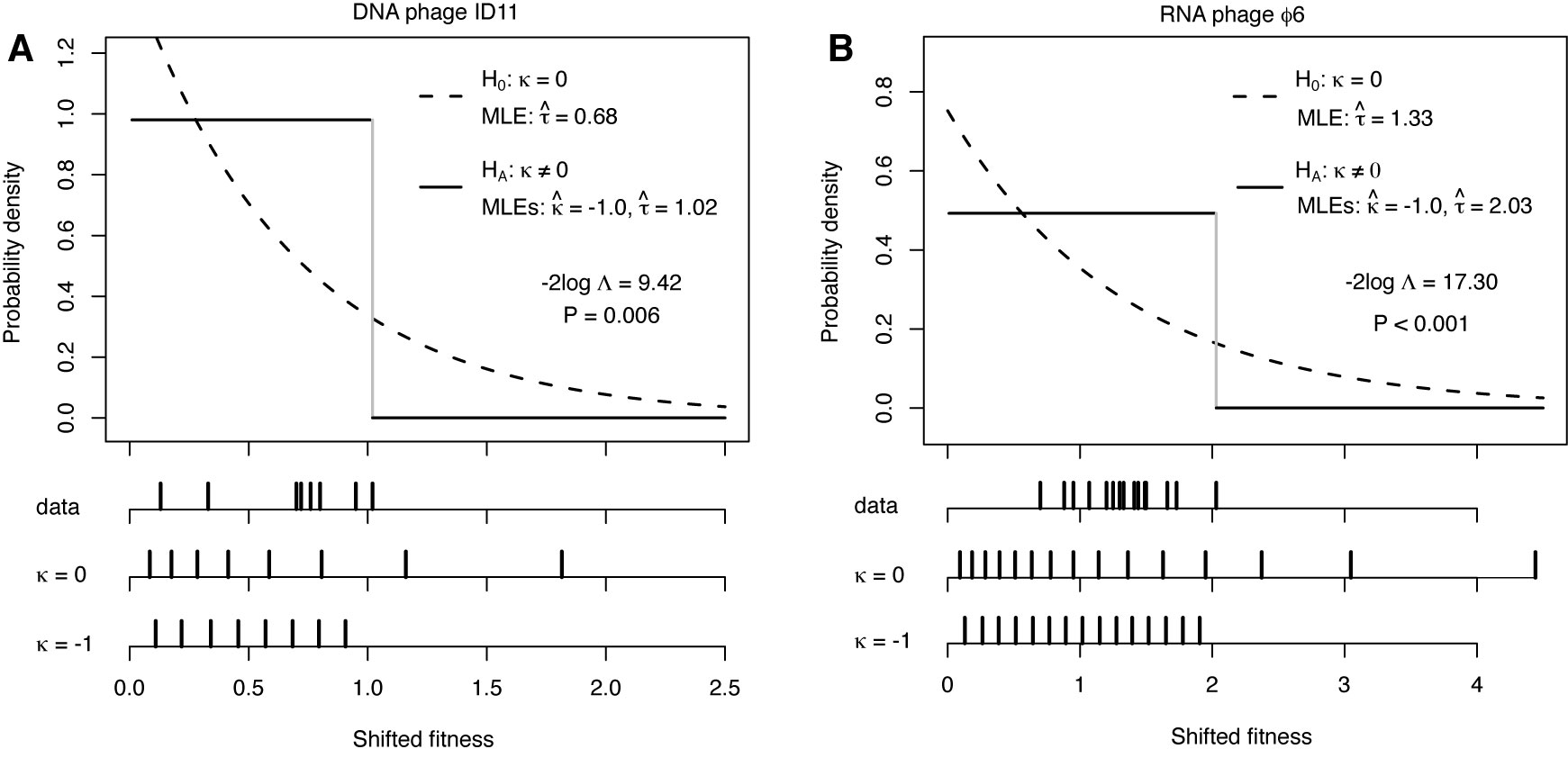 The figure above is from Rokyta et al. (2008), listed in the publications section. Note that the parameter kappa is the same parameter described above. Often, microbial systems can be used to investigate evolutionary phenomena that at first glance are irrelevant to such simple organisms if focus is placed on the basic underlying genetic causes of the phenomena. For example, the very concept of species in microbes is problematic (but see below), yet microbes can still be used to study potential causes of speciation in more complex organisms. Conflict between genes in hybrids is thought to facilitate speciation, and we investigated this possibility using recombinant bacteriophages. A reciprocal cross between two distinct phage genotypes was performed, which precisely exchange their alleles of the major capsid protein. We studied the nature of genic incompatibilities in hybrids by determining the fitness cost of hybridization and by allowing the hybrids to recover fitness in the lab. The types of mutations involved in recovery provided information on the nature of the incompatibilities. The molecular basis for the reduced hybrid fitness is currently being investigated, as are the roles of genetic divergence between genotypes and the particular gene selected for exchange. Environmental Viral Evolution
Snake Venom Evolution
|
 Experimental microbial evolution provides fine-scale information about molecular evolution under controlled, defined conditions. Even though it occurs on a short time scale,
the amount of evolution that can be observed in the lab is limited, and we are therefore limited to primarily adaptive evolution. Furthermore, the selective pressures are simple and constant.
Natural populations of bacteriophages are a largely untapped reservoir of genetic and evolutionary data. Bacteriophages are incredibly abundant in nature, and their simplicity and
small genome size make them particularly suitable for studying genome-wide patterns of molecular evolution.
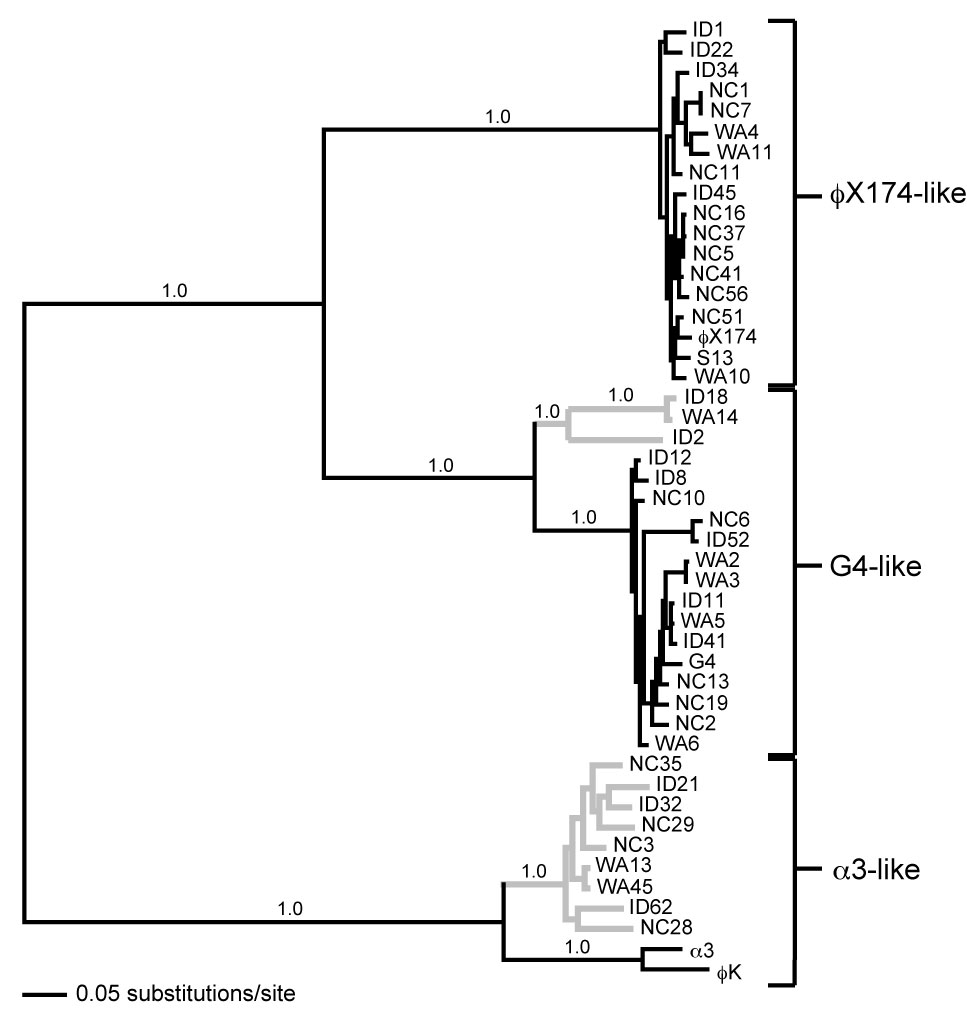
On the left is the phylogeny based on full genome sequences of the 47 characterized microvirid bacteriophages that infect Escherichia coli , described by Rokyta et al. (2006) in the
Experimental microbial evolution provides fine-scale information about molecular evolution under controlled, defined conditions. Even though it occurs on a short time scale,
the amount of evolution that can be observed in the lab is limited, and we are therefore limited to primarily adaptive evolution. Furthermore, the selective pressures are simple and constant.
Natural populations of bacteriophages are a largely untapped reservoir of genetic and evolutionary data. Bacteriophages are incredibly abundant in nature, and their simplicity and
small genome size make them particularly suitable for studying genome-wide patterns of molecular evolution.
On the left is the phylogeny based on full genome sequences of the 47 characterized microvirid bacteriophages that infect Escherichia coli , described by Rokyta et al. (2006) in the
 Experimental microbial evolution provides high resolution data on adaptive molecular evolution but only under highly simplified conditions. The Rokyta lab is now studying the population genetics
and phylogenetics of venom genes from a variety of snake species. We are particularly interested in measuring the strength of selection acting on venom genes and determining how gene flow and
selection interact in a geographic context to result in local adaptation. We are currently sequencing venom genes from several species by means of venom-gland transcriptomics
(see the
Experimental microbial evolution provides high resolution data on adaptive molecular evolution but only under highly simplified conditions. The Rokyta lab is now studying the population genetics
and phylogenetics of venom genes from a variety of snake species. We are particularly interested in measuring the strength of selection acting on venom genes and determining how gene flow and
selection interact in a geographic context to result in local adaptation. We are currently sequencing venom genes from several species by means of venom-gland transcriptomics
(see the