Version 1.0
![]()
©Kent Vander Velden and Gavin Naylor
Iowa State University
| Email: gnaylor@iastate.edu graphix@iastate.edu |
Address: Gavin Naylor Laboratory Department of Zoology and Genetics Science II Iowa State University 50011 IA USA |
Tel:
515-294-4257 Fax: 515-294-8457 |
Motivation: We anticipate that this software will be especially useful for biologists wishing to understand the relationship between evolutionary change in sequence and any concommittent change that occurs in structure. As more data become avaialble it will also be able to investigate whether there exist trajectories of shape change over evolutionary time. |
| Principle:
It is based upon ideas of
Geometric morphometrics originally conceived by Bookstein and Rohlf
to compare anatomical structures. Amino acid sequences are first
aligned using a multiple alignment. This provides the hypothesis for
homology (correspondence) among the residues of different proteins.
The 3-D coordinates of each residue are then used as input data for
the program. Structures are translated, rotated and size
standardized to a common reference. The software then conducts an
eigen decomposition of the shape differences between structures as a
series of "partial warps" (based on a bending energy matrix
generated from superimposing one structure onto another)
|
| Demo:
Movies for the two previously described features are
available. (1) 3-D
Morphometry (Cubes) (2) 3-D
Morphometry (Myoglobin) | ||||||
| ||||||
Features: |



| Features: |
|
2-D morphometry: A particularly powerful aspect of the geometric morphometric
approach is the fact that it embodies a reciprocal relationship
between a point in a shape space and a three dimensional
representation of a structure. |
 |
| 3-D morphometry:
Comparisons between protein structures generates a high
dimensional shape space within which Procrustes Distance provides a
similarity metric that closely corresponds to a natural sense of
overall shape similarity. Projections from the shape space allow us
to visualize shape similarity as an ordination on a plane.
|
 |
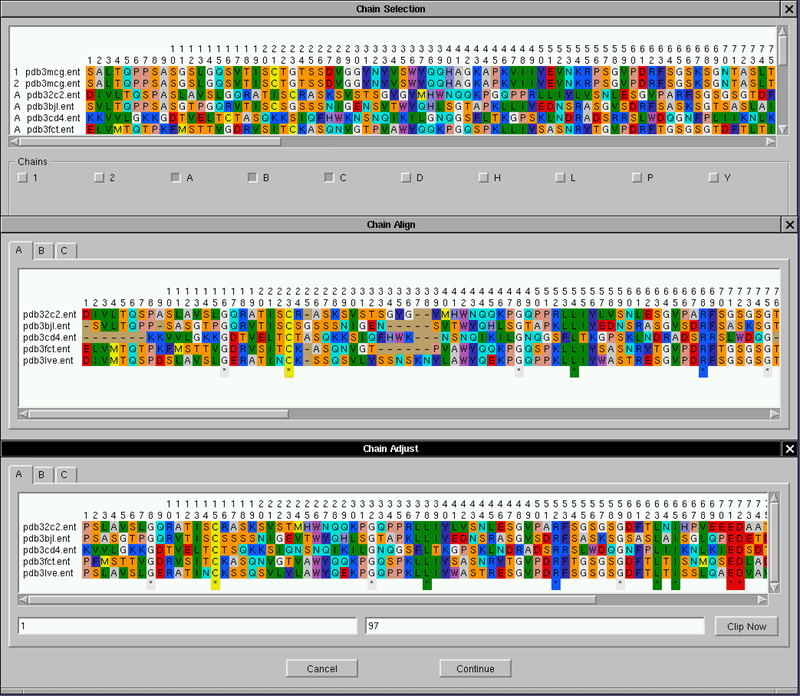
| 3-D morphometry PDB parser:
This feature allows us to aligne the data sequences and the structure from a PDB file. |
 |
| Availability:
|
Copyright © 2001, Iowa State University, all rights reserved.